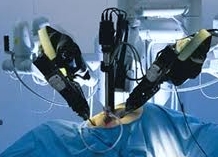


According to the FDA, doctors should stop using a procedure performed on thousands of American women a year in the course of hysterectomy uterine surgery, because it increases the risk of spreading cancerous tissue.
 The procedure, power morcellation, cuts uterine tissue into pieces that can be pulled out through the incisions made during laparoscopic surgery. The morcellator devices, have been widely used in laparoscopic operations to remove fibroid tumors from the uterus, or to remove the entire uterus.
The procedure, power morcellation, cuts uterine tissue into pieces that can be pulled out through the incisions made during laparoscopic surgery. The morcellator devices, have been widely used in laparoscopic operations to remove fibroid tumors from the uterus, or to remove the entire uterus.
Morcellator Devices: No Clinical Trials